Exploring the realm of Clinical Trials and Drug Approval Process, this introduction delves into the intricacies of medical research and regulatory processes, providing a comprehensive overview for readers in a engaging and informative manner.
From the initial phases of clinical trials to the final approval of new drugs, this topic sheds light on the vital steps and considerations involved in bringing medications to market.
CLINICAL TRIALS

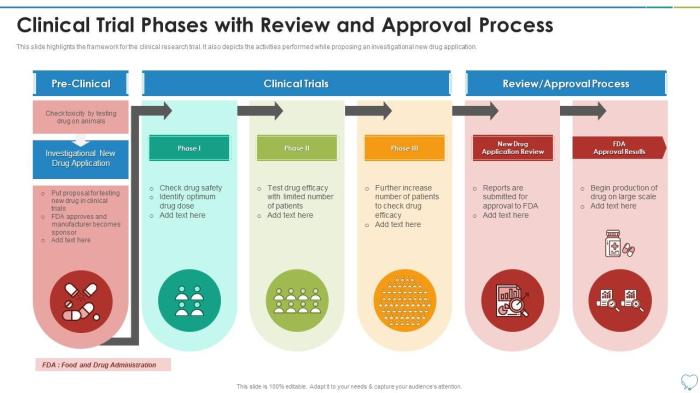
Clinical trials play a crucial role in the drug approval process by evaluating the safety and effectiveness of new medications before they can be approved for public use. These trials help researchers gather data on the drug’s effects on humans, ensuring that it meets regulatory standards and is safe for patients.
Different Phases of Clinical Trials
Clinical trials are typically conducted in four phases:
- Phase 1: Involves testing the drug on a small group of healthy volunteers to determine its safety, dosage range, and potential side effects.
- Phase 2: Expands the study to a larger group of individuals with the targeted disease or condition to assess the drug’s effectiveness and further evaluate its safety.
- Phase 3: Involves a larger group of participants to confirm the drug’s effectiveness, monitor side effects, and compare it to existing treatments.
- Phase 4: Occurs after the drug has been approved and involves post-market surveillance to monitor its long-term safety and effectiveness.
Significance of Randomized Controlled Trials
Randomized controlled trials (RCTs) are considered the gold standard in clinical research due to their ability to minimize bias and provide reliable evidence on a drug’s efficacy. In RCTs, participants are randomly assigned to either the treatment group receiving the drug or the control group receiving a placebo or standard treatment, allowing researchers to compare outcomes accurately.
Comparison of Observational Studies and Experimental Studies
- Observational Studies: These studies observe participants in their natural settings without any intervention from researchers. They can help identify potential associations between variables but do not establish causation.
- Experimental Studies: Also known as interventional studies, these trials involve actively intervening by administering the drug or treatment to participants. They are designed to establish causation and determine the effectiveness of the intervention.
DRUG DEVELOPMENT PROCESS
Developing a new drug involves a series of rigorous steps to ensure its safety and efficacy before it can be approved for use in patients. The process typically includes preclinical studies, clinical trials, regulatory approval, and post-market monitoring.
Role of Preclinical Studies
Preclinical studies play a crucial role in drug development by assessing the safety and efficacy of a potential new drug before it is tested in humans. These studies are conducted in vitro (in the lab) and in vivo (in animals) to gather preliminary data on the drug’s pharmacokinetics, toxicology, and potential side effects.
- Preclinical studies help researchers identify the most promising drug candidates and optimize their dosage and formulation.
- These studies also provide valuable information on the drug’s mechanism of action and potential risks, guiding the design of clinical trials.
- Results from preclinical studies are submitted to regulatory agencies as part of the drug approval process, demonstrating the drug’s safety profile and potential benefits.
Importance of Toxicity Testing
Toxicity testing is a critical component of drug development as it helps identify any potential harmful effects of a new drug on living organisms. This testing is essential for ensuring the safety of patients who may eventually use the drug.
Understanding the toxicological profile of a drug is crucial in determining its therapeutic window and potential side effects.
- Toxicity testing involves assessing the drug’s effects on various organs and systems in the body, as well as its potential to cause genetic mutations or carcinogenicity.
- By evaluating the drug’s toxicity in preclinical studies, researchers can make informed decisions about its safety and dosage levels for human trials.
- Regulatory agencies require comprehensive toxicity data as part of the drug approval process to assess the risk-benefit ratio of the new drug.
Timelines of Drug Development
The timelines for developing a new drug can vary depending on the type of medication and the complexity of the disease it aims to treat. Generally, the drug development process can take several years, with distinct phases and milestones along the way.
For example, the development of a new cancer drug may involve extensive preclinical studies, multiple phases of clinical trials, and regulatory review, leading to a longer timeline compared to a new antibiotic for a common infection.
- Fast-track programs and expedited reviews may accelerate the development timeline for certain drugs, especially those targeting unmet medical needs or rare diseases.
- Overall, the drug development process requires careful planning, collaboration among stakeholders, and adherence to regulatory standards to ensure the safe and timely introduction of new therapies to patients.
REGULATORY AGENCIES
Regulatory agencies play a crucial role in the drug approval process to ensure the safety and efficacy of pharmaceutical products before they reach the market.
Key Regulatory Agencies
- The Food and Drug Administration (FDA): The FDA is the regulatory agency responsible for approving drugs in the United States. It evaluates the safety and efficacy of new drugs through rigorous clinical trials and reviews data submitted by pharmaceutical companies.
- The European Medicines Agency (EMA): The EMA is the regulatory agency for drug approval in the European Union. It follows similar processes to the FDA in evaluating the safety and efficacy of new drugs.
- Other Regulatory Bodies: Other countries have their regulatory agencies, such as Health Canada, the Pharmaceuticals and Medical Devices Agency (PMDA) in Japan, and the Therapeutic Goods Administration (TGA) in Australia.
Differences in Drug Approval Processes
- The FDA typically requires more extensive clinical data compared to the EMA, resulting in longer approval timelines in the U.S. However, the EMA may have more centralized decision-making processes for drug approvals across European countries.
- Regulatory agencies in different countries may have varying requirements for clinical trial design, endpoints, and data analysis, leading to differences in drug approval processes.
Criteria for Evaluating Drug Efficacy and Safety
- Regulatory agencies evaluate drug efficacy based on clinical trial data, including the demonstration of significant treatment effects compared to a control group or standard of care.
- Drug safety is assessed by monitoring adverse events reported during clinical trials and considering the benefit-risk profile of the drug for patients.
Role of Post-Market Surveillance
Post-market surveillance involves monitoring approved drugs once they are available to the public to identify any new safety concerns or adverse effects that may not have been evident during clinical trials. Regulatory agencies work closely with healthcare providers, patients, and pharmaceutical companies to ensure the ongoing safety and effectiveness of approved drugs.
ETHICAL CONSIDERATIONS
In the realm of clinical trials, ethical considerations play a crucial role in ensuring the safety, rights, and well-being of participants. It is essential to adhere to strict ethical standards to uphold the integrity of the research process and protect the rights of individuals involved.
Ethical Standards in Clinical Trials
- Researchers must obtain informed consent from all participants before they can enroll in a clinical trial. This process ensures that participants are fully aware of the risks, benefits, and procedures involved in the study.
- Confidentiality of participants’ personal information must be maintained throughout the trial to protect their privacy and data security.
- Researchers should disclose any potential conflicts of interest that may influence the study outcomes to maintain transparency and integrity.
Importance of Informed Consent
Informed consent is a critical component of ethical clinical research as it empowers participants to make well-informed decisions about their involvement in a trial. By providing detailed information about the study objectives, procedures, potential risks, and benefits, researchers ensure that participants can voluntarily consent to participate without coercion or manipulation.
Ensuring Patient Safety in Trials
To safeguard the well-being of participants, researchers must closely monitor and evaluate the safety and efficacy of the investigational drug or treatment throughout the trial. Any adverse events or side effects must be promptly reported and addressed to protect the health of participants.
Role of Institutional Review Boards (IRBs)
Institutional Review Boards (IRBs) serve as independent ethics committees responsible for reviewing, approving, and monitoring clinical trials to ensure that they comply with ethical standards and regulatory requirements. IRBs evaluate the study protocol, informed consent process, participant recruitment, and ongoing safety monitoring to protect the rights and welfare of trial participants.
In conclusion, the journey from clinical trials to drug approval is a complex yet essential process that ensures the safety and efficacy of medications. By understanding the roles of regulatory agencies, ethical considerations, and research protocols, we gain insight into the meticulous path to medication approval.
FAQ Summary
What are the different phases of clinical trials?
The phases include testing the drug on a small group of people, then a larger group, and finally comparing it with existing treatments.
How do regulatory agencies evaluate drug safety and efficacy?
Regulatory agencies assess data from clinical trials, preclinical studies, and post-market surveillance to ensure drugs are safe and effective.
Why is informed consent important in clinical research?
Informed consent ensures that participants understand the risks and benefits of participating in a clinical trial before they agree to take part.
What is the role of Institutional Review Boards (IRBs) in clinical trials?
IRBs oversee the ethical conduct of clinical trials, protecting the rights and welfare of participants throughout the research process.




